Fda Investigator Brochure Guidance
Fda Investigator Brochure Guidance - Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. This section provides guidance to investigators and sponsors (i.e., the responsible parties) on appropriate management of data integrity, traceability and security, thereby allowing the. It acts as a key. The investigator's brochure for medical devices provides crucial information specific to the study and usage of medical devices in clinical settings. Guidance documents are available from fda. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects. The kind, duration, and scope of animal and other tests required varies with the duration and nature of the proposed clinical investigations. For the most recent version of a guidance, check the fda guidance web page at. What is the statement of investigator, form fda 1572? Gmp regulatory intell.150 docs added each monthover 14k searchable 483s Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. It acts as a key. Fda developed this guidance in response to concerns 21 raised by the ire3 community, including concerns raised at a march 2005 public hearing2, that 22 increasingly large volumes. For the most recent version of a guidance, check the fda guidance web page at. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects. The goal of this guidance is to help investigators better meet their responsibilities with respect to protecting human subjects and ensuring the integrity of the data from clinical. This guidance clarifies requirements for data and data presentation in 21 cfr 312.22 and 312.23 related to the initial entry into human studies in the united states of an investigational drug. The kind, duration, and scope of animal and other tests required varies with the duration and nature of the proposed clinical investigations. The statement of investigator, form fda 1572 (1 572), is an agreement signed by the investigator to provide certain information to. Gmp regulatory intell.150 docs added each monthover 14k searchable 483s Gmp regulatory intell.150 docs added each monthover 14k searchable 483s Providing investigators with the necessary information to. In drug development, the investigator’s brochure (ib) summarises the main elements of the entire development programme to date, primarily for the benefit of investigators conducting clinical. 50 finalized, the 2012 final guidance continues to represent fda’s current thinking about 51 investigators’ responsibilities for. Gmp regulatory intell.150 docs added each monthover 14k searchable 483s Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. What is the statement of investigator, form fda 1572? In drug development, the investigator’s brochure (ib) summarises the main elements of the entire development programme. This guidance clarifies requirements for data and data presentation in 21 cfr 312.22 and 312.23 related to the initial entry into human studies in the united states of an investigational drug. The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a sample consent. Guidance documents are available from fda.. This guidance clarifies requirements for data and data presentation in 21 cfr 312.22 and 312.23 related to the initial entry into human studies in the united states of an investigational drug. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects.. Fda developed this guidance in response to concerns 21 raised by the ire3 community, including concerns raised at a march 2005 public hearing2, that 22 increasingly large volumes. 50 finalized, the 2012 final guidance continues to represent fda’s current thinking about 51 investigators’ responsibilities for safety reporting for inds and ba/be studies. The investigator’s brochure (ib) is a compilation of. This guidance clarifies requirements for data and data presentation in 21 cfr 312.22 and 312.23 related to the initial entry into human studies in the united states of an investigational drug. The goal of this guidance is to help investigators better meet their responsibilities with respect to protecting human subjects and ensuring the integrity of the data from clinical. It. This section provides guidance to investigators and sponsors (i.e., the responsible parties) on appropriate management of data integrity, traceability and security, thereby allowing the. The goal of this guidance is to help investigators better meet their responsibilities with respect to protecting human subjects and ensuring the integrity of the data from clinical. Fda developed this guidance in response to concerns. Guidance documents are available from fda. This guidance clarifies requirements for data and data presentation in 21 cfr 312.22 and 312.23 related to the initial entry into human studies in the united states of an investigational drug. Fda developed this guidance in response to concerns 21 raised by the ire3 community, including concerns raised at a march 2005 public hearing2,. It acts as a key. The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a sample consent. For the most recent version of a guidance, check the fda guidance web page at. The statement of investigator, form fda 1572 (1 572), is an agreement signed by the investigator to. The goal of this guidance is to help investigators better meet their responsibilities with respect to protecting human subjects and ensuring the integrity of the data from clinical. What is the statement of investigator, form fda 1572? This guidance clarifies requirements for data and data presentation in 21 cfr 312.22 and 312.23 related to the initial entry into human studies. The statement of investigator, form fda 1572 (1 572), is an agreement signed by the investigator to provide certain information to. What is the statement of investigator, form fda 1572? The kind, duration, and scope of animal and other tests required varies with the duration and nature of the proposed clinical investigations. Guidance documents are available from fda. Owing to the importance of the ib in maintaining the safety of human subjects in clinical trials, and as part of their guidance on good clinical practice (gcp), the u.s. Providing investigators with the necessary information to. 50 finalized, the 2012 final guidance continues to represent fda’s current thinking about 51 investigators’ responsibilities for safety reporting for inds and ba/be studies. For the most recent version of a guidance, check the fda guidance web page at. This guidance clarifies requirements for data and data presentation in 21 cfr 312.22 and 312.23 related to the initial entry into human studies in the united states of an investigational drug. The investigator's brochure for medical devices provides crucial information specific to the study and usage of medical devices in clinical settings. It acts as a key. The goal of this guidance is to help investigators better meet their responsibilities with respect to protecting human subjects and ensuring the integrity of the data from clinical. Fda developed this guidance in response to concerns 21 raised by the ire3 community, including concerns raised at a march 2005 public hearing2, that 22 increasingly large volumes. In drug development, the investigator’s brochure (ib) summarises the main elements of the entire development programme to date, primarily for the benefit of investigators conducting clinical. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects.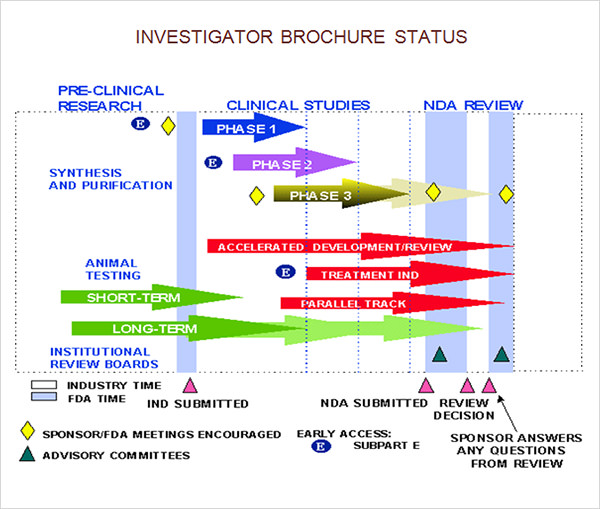
FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

Investigator Brochure Template Fda

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

PPT ICHGCP & FDA Regulations Differences PowerPoint Presentation

Investigator Brochure Template Fda

Investigator Brochure Template Fda

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word
The Documents Reviewed Should Include The Complete Documents Received From The Clinical Investigator, Such As The Protocol, The Investigator's Brochure, A Sample Consent.
Good Clinical Practice (Gcp) Is An International Ethical And Scientific Quality Standard For Designing, Conducting, Recording And Reporting Trials That Involve The Participation Of Human.
This Section Provides Guidance To Investigators And Sponsors (I.e., The Responsible Parties) On Appropriate Management Of Data Integrity, Traceability And Security, Thereby Allowing The.
Gmp Regulatory Intell.150 Docs Added Each Monthover 14K Searchable 483S
Related Post: